

How do you use a virtual cell to do something actually useful? (1/3)
Identifying anti-PD-1 responders

Note: this is part of a series of three posts discussing how the therapeutics team at Noetik have used our virtual cell model, OCTO-VC, for practically useful, therapeutics-relevant tasks. The Introduction section will stay the same for each one, skip down to the next section if you’ve already read one of these before.
Part 1: Identifying anti-PD-1 responders
Part 2: Refining clinical trial eligibility to the right subgroups
Part 3: Virtual perturbations that shift T cell effector state in humans
Table of contents:
Introduction
A lot of people have been very interested in ‘virtual cells’ lately. An exact definition is difficult to find, but one offered by a recent Cell perspective paper is the following:
Our view of [a virtual cell] is a learned simulator of cells and cellular systems under varying conditions and changing contexts, such as differentiation states, perturbations, disease states, stochastic fluctuations, and environmental conditions. In this context, a virtual cell should integrate broad knowledge across cell biology. [Virtual cells] must work across biological scales, over time, and across data modalities and should help reveal the programming language of cellular systems and provide an interface to use it for engineering purposes.
It’s an exciting idea! A computational simulation of a cell should be, theoretically, exceedingly useful for all sorts of clinical and preclinical research, by virtue of being able to eschew expensive wet-lab efforts in favor of cheaper (and potentially more reliable) GPU time. So it is no surprise that a great deal of research is already being actively done in this area. Elliot Hershberg, a venture capitalist at Amplify Partners, recently compiled a small summary of ongoing work here:

But as with every promised revolution in the life sciences, the revolution will hesitantly admit some nuances upon questioning.
Of highest concern is the fact that nearly all virtual cell model efforts being worked on are are not virtual cells of human biology, but rather cancer cell lines, which—while convenient, well-characterized, and infinitely malleable—are far from the true physiological complexity of healthy or diseased human tissue. Due to this, figuring out how their insights extend into assisting with the drug development process is usually another hard problem in and of itself. But, to be clear, this doesn’t mean they aren’t useful. Biological research being done on cancer cell lines is a common phenomenon at the preclinical research stage, which is what nearly all virtual cell models are currently geared towards assisting.
This partially answers the question why, despite how exciting ‘virtual cells’ seem, there are very few, clear-cut examples of how such methods will be ultimately used. That vagueness is partly built into the reality of early-stage biology, so it’ll be years before the ultimate impact of this line of research is felt.
But one area of virtual cells that could have a concrete value-add in the immediate short-term is the deployment of them at the clinical stage of drug development. After all, this is where the real bottlenecks lie: trials are slow, expensive, and fraught with uncertainty, and even small improvements here can ripple into huge downstream gains. Of course, while the opportunity here is massive, the downside of touching this area is that it is hard to do. Very, very hard. As a result, there is almost no virtual cell effort meant to operate at the clinical stages of drug development, even though the translation problem there is, theoretically, ‘easy’.
Other than us. Noetik is building virtual cells with the explicit goal of assisting with clinical-stage problems: identifying responders to drugs and refining patient inclusion criteria for trials. At the same time, we believe that the tools we create in this process will also have powerful applications in pivotal, high-risk areas of preclinical research, such as target selection, while remaining grounded in human-level data. All three will be discussed in this essay series.
How do we do this? Our view is simple: the shortest path to usefulness is not maximal simulation on unrealistic biology, but grounded observations into realistic biology. We built that foundation first. Every datapoint that trains our virtual cell models comes from human tumor resections: 77M cells across ~2,500 patients across a dozen+ cancers, with paired spatial transcriptomics, spatial proteomics, exomes, and H&E’s from each one collected in our lab. In total, this is easily one of the largest datasets of its kind. And not a single cell line. We strongly believe that this means the path from in-silico workflows to something clearly translatable is far more direct: human to human, rather than detouring through unrealistic animal or cell models.
That difference matters! In cancer, translation is the bottleneck. Drugs fail, not because they don’t work in preclinical settings, but because they don’t work in real human patients.
Using this human-derived tumor data, one of the virtual cell models we’ve created is ‘OCTO-VC’. This model is entirely trained on 1000-plex spatial transcriptomes, and its core task is deliberately prosaic: given the transcriptome of a few neighboring cells, reconstruct the “center cell” transcriptome—over every cell, in every tumor, for every patient. We released a (very long) post late last year discussing it in depth for those who are curious about the machine-learning details, alongside an online demo.
But what wasn’t discussed in that earlier post is how one can use models like this for clinically meaningful, non-trivial problems.
In this essay series, we hope to do exactly that, by showing three case studies of times where OCTO-VC was directly useful for our therapeutics team. This is part 1, which will discuss how we used the model to find true anti-PD-1 responders inside PD-L1–positive cohorts.
Identifying anti-PD-1 responders
Therapeutic Context:
One of the most common (and effective) therapies in cancer are anti-PD-1 drugs. The underlying biology is straightforward: many tumor cells express PD-L1 on their surface, which binds PD-1 receptors on T cells to dampen T-cell activity. Anti-PD-1 (or anti-PD-L1) antibodies block this inhibitory interaction, allowing T cells to attack the tumor. But not all cancers rely on this pathway. Some tumors have little to no PD-L1 expression, meaning that drugs operating on that mechanism would, in principle, have limited effect. This has led to a common clinical rule of thumb: patients are considered potential candidates for anti-PD-1/PD-L1 therapy if ≥1% of their tumor cells express PD-L1, or are PD-L1+. But this still isn’t perfect. Even with this inclusion metric, roughly half of patients still do not respond to this therapy, even if they are PD-L1+, and it is unclear why.
Question:
Can OCTO-VC improve how well we can identify responders to this drug?
What we found:
Seeing how well OCTO-VC can help us here is quite straightforward: create a high-dimensional embedding of each of our tumor cores that we have responder data for, and see if the embeddings of responders differ from those of non-responders. And, most importantly, is the clustering better than a good baseline, as in, the usual patient inclusion criteria?
You may be instinctively surprised by the fact that OCTO-VC’s value here doesn’t come from the usual virtual cell trick of simulating perturbations, but instead from the far simpler act of representation. But this is, in fact, the most reliable way to rely on models like this; it allows our underlying, extremely rich data to ‘speak for itself’ without needing human intervention.
Using a small cohort of patients—15 responders and 24 non-responders, with both groups meeting the “ideal candidate” criterion above, or PD-L1 tumor proportion score ≥1%—we generated an embedding for each of their tumors using OCTO-VC. The below graph shows the embeddings for all our core samples, reduced to two dimensions via PCA. The ones we have PD-1 responder data for are colored in either green or magenta.
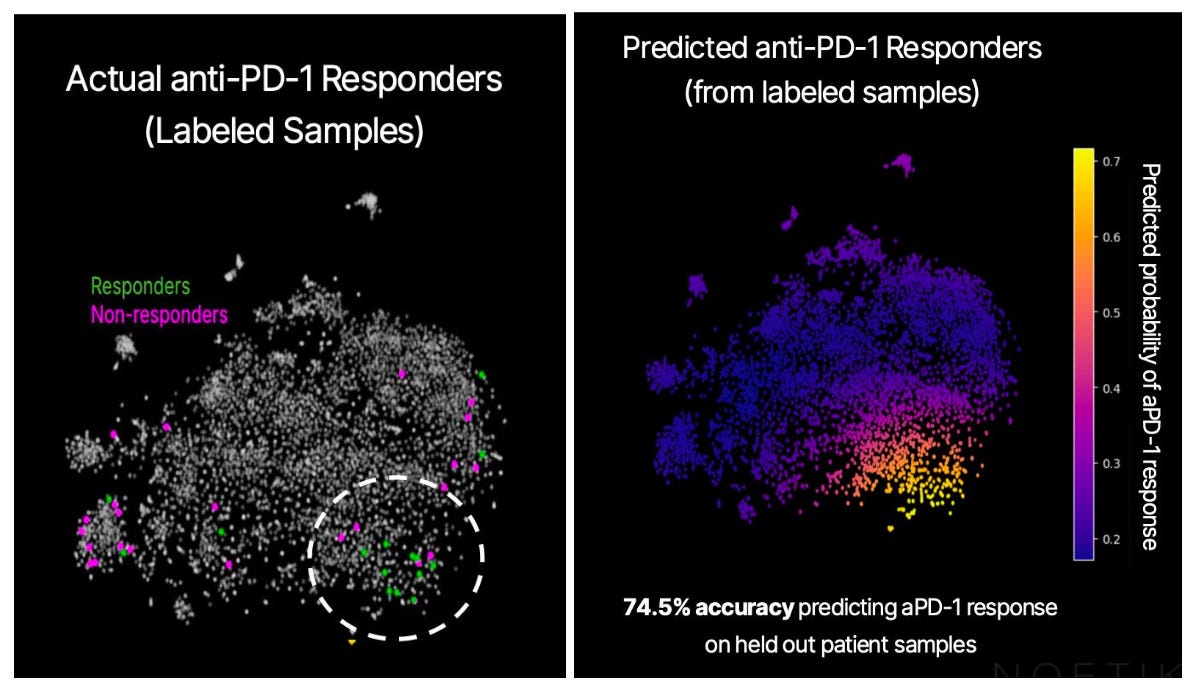
The responders seem to mostly be in the lower right quadrant, so there’s meaningful separation in the entirely unsupervised embedding. And, training a basic model on the PCA reduction allows us to quantify the signal, showing that predictions match up well with the response cluster, and that it is above chance. Here is the associated confusion matrix of the trained model:
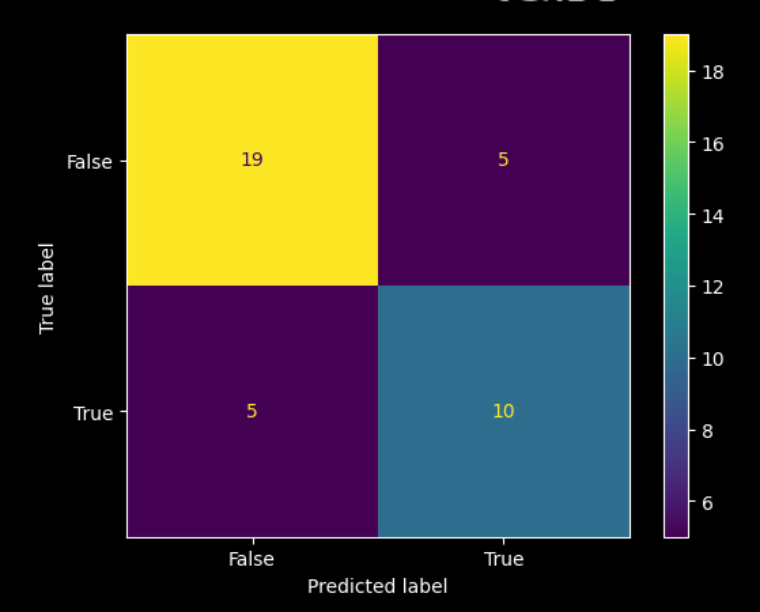
Remember, we’re working off a pre-selected patient population here. If “1% of tumor cells expressing PD-L1” were a really good biomarker with no real room left to improve upon, we wouldn’t be able to further subdivide the likely-to-respond patient population any further. The fact that we’re able to easily spot the “response cluster” in the embedding space is encouraging to us, and implies that OCTO-VC is capturing response-relevant biology that the 1% rule misses.
Future Directions:
The cancer field has been through a lot of definitions on what is the ‘most’ important factor to care about regarding a tumor. At first, it was about histology. Lung cancer could be separated into small-cell, squamous, non-small-cell, and so on. Then arrived the genetic era, when EGFR mutations or ALK fusions could, by themselves, dictate treatment. Now we are in the protein marker era, with PD-L1 expression being the most commonly deployed stratifier for checkpoint blockade.
None of these were wrong, but each was only capturing a small fragment of the whole.
Tumors are not uniform entities, but rather shifting ecosystems of cells, pathways, and immune structures. Understanding this complexity, to a large extent, may be beyond human intuition or comprehension. We at Noetik strongly believe that machine intelligence is the only way to grasp the tumor microenvironment in full.
The building of OCTO-VC, and the fact that anti-PD1 responders are so clearly separated in its embedding space, implies to us that this conviction is directionally accurate. Also, since the underlying data is all sourced from human tumors, we can easily pin down what other biological features predicted anti-PD-1 responders correlate with, both to reassure ourselves that they make sense and that they can be converted to usable assays. And indeed, both are present: CD8 infiltration, high interferon gamma levels, and antigen presentation markers (to name a few) align with responder status.
Of course, the real significance here is not in what we can do for anti-PD1 therapies—many people have worked on this exact subject before—but rather, in how easily our methodology can be extended to any arbitrary cancer drug. In other words, if OCTO-VC can isolate a subset of checkpoint responders from within an already enriched PD-L1+ population, then it should also be able to refine other trial cohorts. Our first partnership is with Agenus, a public biotechnology company, to see if our model is capable of accurately distinguishing between responders/non-responders from a recent clinical trial that Agenus ran. We’re looking forward to reporting what our results are here!
If any of this seems interesting, and you’d like to chat further, please reach out to info@noetik.ai!
Of course, one won’t always have response data. We think there are useful ways that OCTO-VC can be used in those situations as well, which is something we’ll discuss in part 2 of this series, covering how our model can be used for expanding eligibility in clinical trial design even when lacking access to true response/non-response data.