

How do you use a virtual cell to do something actually useful? (2/3)
Refining clinical trial eligibility to the right subgroups

Note: this is part two of a series of three posts discussing how the therapeutics team at Noetik have used our virtual cell model, OCTO-VC, for practically useful, therapeutics-relevant tasks. The Introduction section will stay the same for each one, skip down to the next section if you’ve already read one of these before.
Part 1: Identifying anti-PD-1 responders
Part 2: Refining clinical trial eligibility to the right subgroups
Part 3: Virtual perturbations that shift T cell effector state in humans
Table of contents:
Introduction
A lot of people have been very interested in ‘virtual cells’ lately. An exact definition is difficult to find, but one offered by a recent Cell perspective paper is the following:
Our view of [a virtual cell] is a learned simulator of cells and cellular systems under varying conditions and changing contexts, such as differentiation states, perturbations, disease states, stochastic fluctuations, and environmental conditions. In this context, a virtual cell should integrate broad knowledge across cell biology. [Virtual cells] must work across biological scales, over time, and across data modalities and should help reveal the programming language of cellular systems and provide an interface to use it for engineering purposes.
It’s an exciting idea! A computational simulation of a cell should be, theoretically, exceedingly useful for all sorts of clinical and preclinical research, by virtue of being able to eschew expensive wet-lab efforts in favor of cheaper (and potentially more reliable) GPU time. So it is no surprise that a great deal of research is already being actively done in this area. Elliot Hershberg, a venture capitalist at Amplify Partners, recently compiled a small summary of ongoing work here:

But as with every promised revolution in the life sciences, the revolution will hesitantly admit some nuances upon questioning.
Of highest concern is the fact that nearly all virtual cell model efforts being worked on are are not virtual cells of human biology, but rather cancer cell lines, which—while convenient, well-characterized, and infinitely malleable—are far from the true physiological complexity of healthy or diseased human tissue. Due to this, figuring out how their insights extend into assisting with the drug development process is usually another hard problem in and of itself. But, to be clear, this doesn’t mean they aren’t useful. Biological research being done on cancer cell lines is a common phenomenon at the preclinical research stage, which is what nearly all virtual cell models are currently geared towards assisting.
This partially answers the question why, despite how exciting ‘virtual cells’ seem, there are very few, clear-cut examples of how such methods will be ultimately used. That vagueness is partly built into the reality of early-stage biology, so it’ll be years before the ultimate impact of this line of research is felt.
But one area of virtual cells that could have a concrete value-add in the immediate short-term is the deployment of them at the clinical stage of drug development. After all, this is where the real bottlenecks lie: trials are slow, expensive, and fraught with uncertainty, and even small improvements here can ripple into huge downstream gains. Of course, while the opportunity here is massive, the downside of touching this area is that it is hard to do. Very, very hard. As a result, there is almost no virtual cell effort meant to operate at the clinical stages of drug development, even though the translation problem there is, theoretically, ‘easy’.
Other than us. Noetik is building virtual cells with the explicit goal of assisting with clinical-stage problems: identifying responders to drugs and refining patient inclusion criteria for trials. At the same time, we believe that the tools we create in this process will also have powerful applications in pivotal, high-risk areas of preclinical research, such as target selection, while remaining grounded in human-level data. All three will be discussed in this essay series.
How do we do this? Our view is simple: the shortest path to usefulness is not maximal simulation on unrealistic biology, but grounded observations into realistic biology. We built that foundation first. Every datapoint that trains our virtual cell models comes from human tumor resections: 77M cells across ~2,500 patients across a dozen+ cancers, with paired spatial transcriptomics, spatial proteomics, exomes, and H&E’s from each one collected in our lab. In total, this is easily one of the largest datasets of its kind. And not a single cell line. We strongly believe that this means the path from in-silico workflows to something clearly translatable is far more direct: human to human, rather than detouring through unrealistic animal or cell models.
That difference matters! In cancer, translation is the bottleneck. Drugs fail, not because they don’t work in preclinical settings, but because they don’t work in real human patients.
Using this human-derived tumor data, one of the virtual cell models we’ve created is ‘OCTO-VC’. This model is entirely trained on 1000-plex spatial transcriptomes, and its core task is deliberately prosaic: given the transcriptome of a few neighboring cells, reconstruct the “center cell” transcriptome—over every cell, in every tumor, for every patient. We released a (very long) post late last year discussing it in depth for those who are curious about the machine-learning details, alongside an online demo.
But what wasn’t discussed in that earlier post is how one can use models like this for clinically meaningful, non-trivial problems.
In this essay series, we hope to do exactly that, by showing three case studies of times where OCTO-VC was directly useful for our therapeutics team.
This is part 2, which will discuss how we would use the model to expand clinical trial eligibility in a real clinical trial.
Refining clinical trial eligibility to the right subgroups
Therapeutic Context:
In our last essay, we talked about how tumor complexity is beyond any human to genuinely grasp, and how we arrived at an understanding of anti–PD-1 response through model embeddings. But in cases when explicit response labels aren’t available, the challenge then shifts. We cannot ask whether responders and non-responders separate. Instead, we must reason through proxy; using machine-learned patterns that recur across tumors that closely correlate with suspected mechanisms of action (MoA) of the drug being studied.
Some background information: as of today, most patient inclusion criteria in cancer clinical trials rely on disturbingly coarse, overly-reductionistic patient inclusion criteria: % of PD-L1 expression across a tumor biopsy (like the previous case study), whether the histology says a tumor is “triple-negative,” or if sequencing shows the presence of a particular mutation. But even when the cancer field explores the value of more complex markers — which oncologists clearly recognize as important! — the published signals are nearly always fragmentary, with a single local motif, and rarely grasp the full neighborhood or architectural context of a tumor microenvironment.
Why is this? Why aren’t markers more complex? Much of it comes down to the fact that the logistics of designing and validating even mildly complex assays are essentially intractable. Every hypothesis requires years of prospective planning, the right tissue samples, and the ability to multiplex the correct set of markers from the start. If an important signal is missed, the entire study has to be restarted. This is to say nothing of biomarkers that are ML-enabled, operating across dozens or hundreds of axes at once, which is virtually never explored.
As a consequence, trial sponsors are forced into the simplest, most reductive criteria, not because they believe those are the best biology, but because they are the only practical levers available within trial timelines.
Question:
One of the things we’ve been most excited about is using OCTO-VC to take previously impractical hypotheses for drug response prediction, and test them out at scale.
The question here requires some extra context and, because we’re actively exploring it, some obfuscation.
Last year, the FDA halted a late-stage cancer clinical trial run by a large biopharma, not because efficacy wasn’t observed, but because, midway through the trial, efficacy was observed only in ‘Subgroup Z’. As a result, this forced the biopharma to submit a protocol amendment to restrict follow-up trials to only be on the Subgroup Z cohort. This is quite a blow to them, since that cohort is a fair bit smaller!
But, as is typical in cancer trials, patients wind up in Subgroup Z due to an extremely coarse biomarker. Theoretically, the drug that was part of this trial is over a well-trodden target, so there should be a much better way to separate out the ideal patient population. But, like we mentioned, doing any sort of large-scale biomarker study would normally require an enormous multi-year biomarker program—prospectively designing assays, collecting new tissue samples, and validating them across multiple sites. That’s the standard, slow way.
With OCTO-VC, we can invert the order of operations. Instead of starting with a hypothesis, locking in the markers, and then waiting years for data to trickle back, we start with the existing atlas of human tumors and ideate on new ways to separate out responder/non-responder patients.
So, our question is: can OCTO-VC come up with better stratification criteria for selecting responders?
What we found:
First, a basic sanity check was met. Subgroup Z cohort is quite distinct in our embedding space; in the graph below, it is the small, bright yellow-green segment on the left.
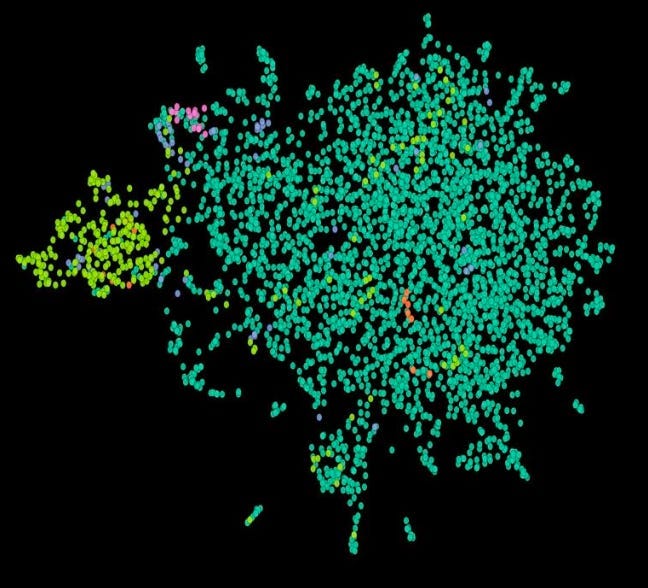
And we knew that that yellow-green segment was filled with responders to the drug. So an easy question to ask OCTO-VC is: what other, more complex marker overlaps with that segment and has a mechanistic rationale for the overlap? After some iterative searching, our therapeutics team found a strong signal: a particular ‘tumor microenvironment concept’ that seems highly enriched in Subgroup Z, but also extends outside of it. While we won’t expand on what the concept is, we believe that it is unlikely to be noise given how biologically relevant it is to the therapy in question.
Here, that concept is shown in the same embedding plot through color; high meaning ‘highly enriched for that concept’:
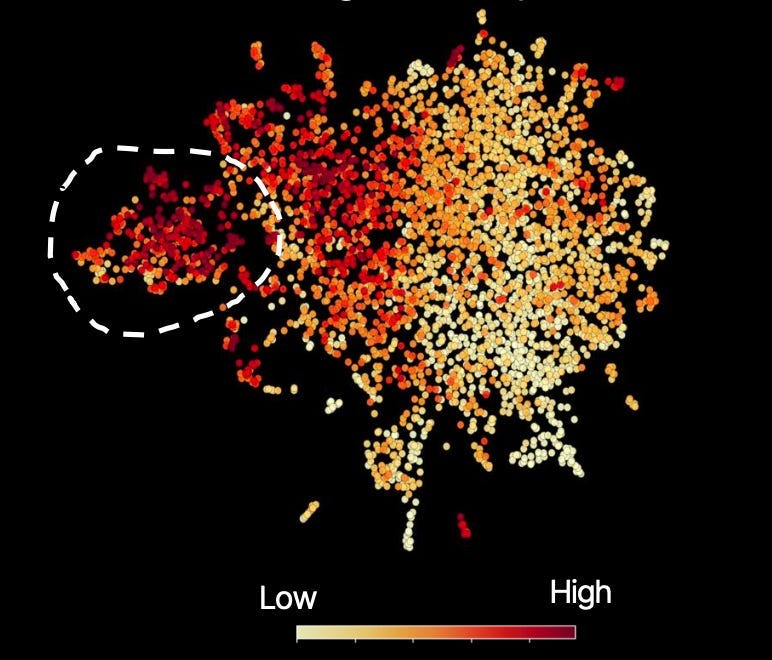
Circled is the true-response cohort, which is high in that concept. But you can notice, slicing through the region outside of Subgroup Z, another large pocket of people high in that concept.
In other words, we believe this biopharma has set overly conservative inclusion criteria. By doing so, they not only leave billions of dollars in potential revenue untapped but, more importantly, will leave an immense number of patients without access to a therapy that has a clear mechanistic reason for meaningfully improving, or even saving, their lives.
Future Directions:
One striking aspect of the OCTO-VC’s embedding space—something that continues to surprise even us—is how clearly it aligns with therapeutic problems, despite having had absolutely no access to perturbational or labeled data. After all, OCTO-VC could separate higher-order cancer definitions (e.g., Subgroup Z vs. not-Subgroup-Z) directly from human tissue, and, with some human judgement, was able to surface MoA-relevant subtypes within them; ones that would be far too costly to ever pull out in the real world. And this phenomenon of ‘clinically meaningful organization’ seems to reoccur across the embedding space!
As an example, basic Leiden clustering of an OCTO-VC embedding space (the same one discussed in this essay, but different from the previous essays PD-1 embedding space) demonstrates tissue-level characteristics that align with therapeutic MoA’s. Annotations here are provided by humans:
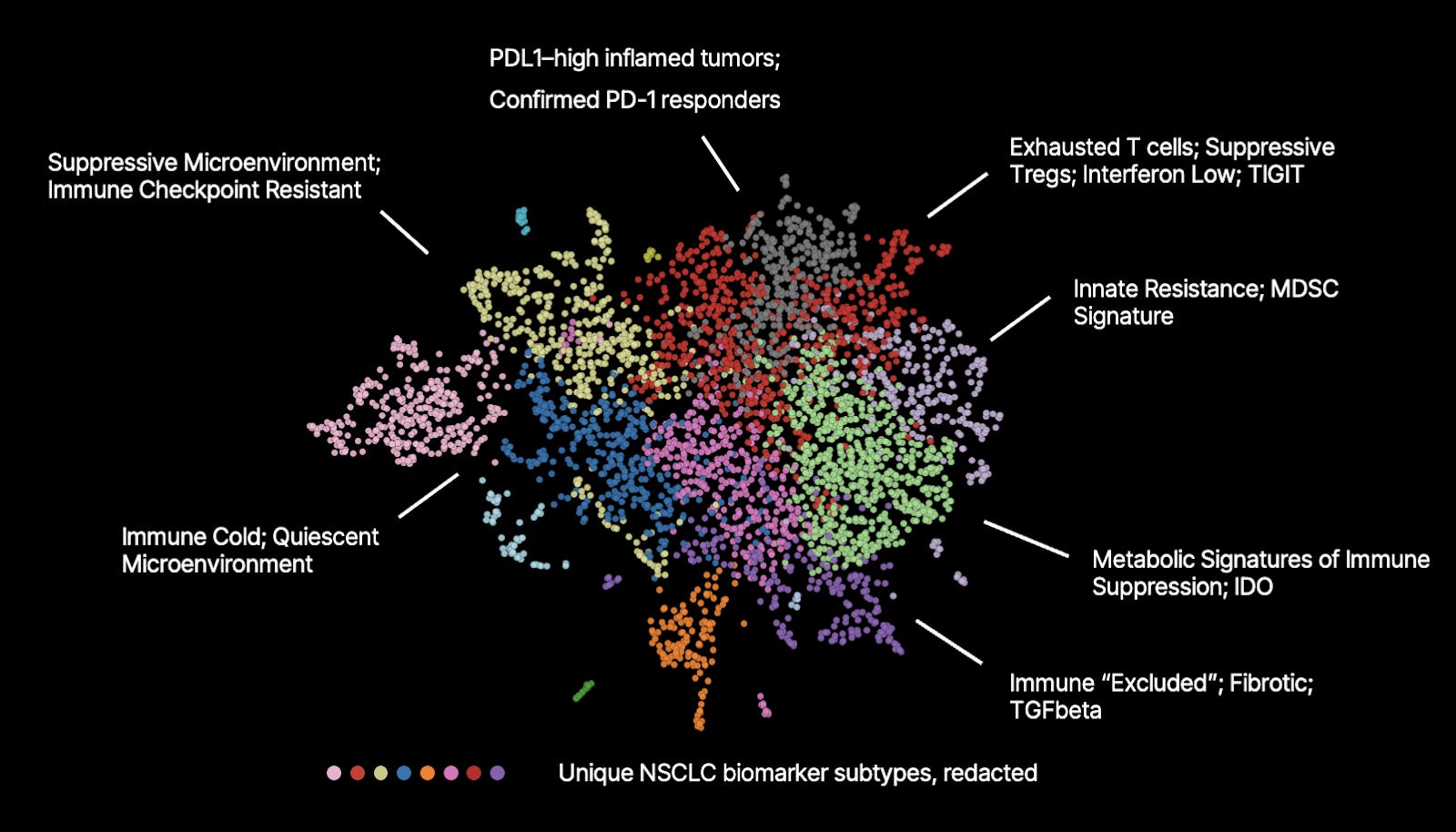
How is this possible? How can the model, without explicit supervision, uncover patterns that map so directly onto biological mechanisms and therapeutic relevance?
One argument is that cancer is a particularly special disease, extremely well-suited to self-supervision tasks. Unlike many other therapeutic areas, oncology has historically been driven by mechanism-based stratification; cancer drugs are often developed and approved not for a broad, undifferentiated population, but for genetically or phenotypically defined subgroups. As a result, the very axes that determine drug response are the same ones that structure our human, tissue-level data. And machine learning is very, very good at dissolving complex, high-dimensional data into those underlying axes.
Of course, turning this work, and others like it, from an analysis into an actual regulatory argument is often another challenge in of itself for many virtual cell efforts. A model can suggest that patients with a certain microenvironmental signature are likely to respond, but to satisfy regulators, that suggestion has to be translated into a practical assay. But this is the core of what makes “virtual cells” particularly useful if they are derived from human data, and not cancer cell lines: this translation is straightforward.
After all, the signatures that OCTO-VC surfaces always have a direct connection to real human tumors. The signatures are often intricate, something that would require years of effort and millions of dollars to define through traditional approaches, but still can be boiled down to a set of measurable markers, morphologies, or local interactions if needed. As an example, the tumor microenvironment concept we discussed above is something that is very amenable to being turned into an assay.
We strongly believe that this ability to create these complex definitions of responder cohort—given only hours of GPU time—can not only expand patient cohorts (as we’ve discussed here), but also rescue otherwise unpromising drugs and open entirely new therapeutic opportunities that were previously invisible under traditional stratification methods.
If this sounds interesting, and you think you’d like to talk further, please reach out to info@noetik.ai!
In our final section, we’ll discuss how even though OCTO-VC is most useful for clinical-stage problems, like patient selection, the same human-tissue-grounded programs also help prioritize targets, without changing the core principle: grounding every insight in real human tissue.
Thank you for this awesome series! Very refreshing to read about virtual cells that have a clear problem statement and concrete applications.
One feature I notice about the white-red heat map is that even in cluster Z, there are ~30% patients who have yellow-orange intensity that is similar to the bulk of the non-responsive cluster. Coupled with the fact that there is red intensity in the non-responsive cluster, could the "concept" you are looking at be adjacent to, but not directly causing, the stratified outcomes? More broadly, how would you "draw the line" in this case of who to include/exclude into certain categories?