

How do you use a virtual cell to do something actually useful? (3/3)
Virtual perturbations that shift T cell effector state in humans

Note: this is part three of a series of three posts discussing how the therapeutics team at Noetik have used our virtual cell model, OCTO-VC, for practically useful, therapeutics-relevant tasks. The Introduction section will stay the same for each one, skip down to the next section if you’ve already read one of these before.
Part 1: Identifying anti-PD-1 responders
Part 2: Refining clinical trial eligibility to the right subgroups
Part 3: Virtual perturbations that shift T cell effector state in humans
Table of contents:
Introduction
A lot of people have been very interested in ‘virtual cells’ lately. An exact definition is difficult to find, but one offered by a recent Cell perspective paper is the following:
Our view of [a virtual cell] is a learned simulator of cells and cellular systems under varying conditions and changing contexts, such as differentiation states, perturbations, disease states, stochastic fluctuations, and environmental conditions. In this context, a virtual cell should integrate broad knowledge across cell biology. [Virtual cells] must work across biological scales, over time, and across data modalities and should help reveal the programming language of cellular systems and provide an interface to use it for engineering purposes.
It’s an exciting idea! A computational simulation of a cell should be, theoretically, exceedingly useful for all sorts of clinical and preclinical research, by virtue of being able to eschew expensive wet-lab efforts in favor of cheaper (and potentially more reliable) GPU time. So it is no surprise that a great deal of research is already being actively done in this area. Elliot Hershberg, a venture capitalist at Amplify Partners, recently compiled a small summary of ongoing work here:

But as with every promised revolution in the life sciences, the revolution will hesitantly admit some nuances upon questioning.
Of highest concern is the fact that nearly all virtual cell model efforts being worked on are not virtual cells of human biology, but rather cancer cell lines, which—while convenient, well-characterized, and infinitely malleable—are far from the true physiological complexity of healthy or diseased human tissue. Due to this, figuring out how their insights extend into assisting with the drug development process is usually another hard problem in and of itself. But, to be clear, this doesn’t mean they aren’t useful. Biological research being done on cancer cell lines is a common phenomenon at the preclinical research stage, which is what nearly all virtual cell models are currently geared towards assisting.
This partially answers the question why, despite how exciting ‘virtual cells’ seem, there are very few, clear-cut examples of how such methods will be ultimately used. That vagueness is partly built into the reality of early-stage biology, so it’ll be years before the ultimate impact of this line of research is felt.
But one area of virtual cells that could have a concrete value-add in the immediate short-term is the deployment of them at the clinical stage of drug development. After all, this is where the real bottlenecks lie: trials are slow, expensive, and fraught with uncertainty, and even small improvements here can ripple into huge downstream gains. Of course, while the opportunity here is massive, the downside of touching this area is that it is hard to do. Very, very hard. As a result, there is almost no virtual cell effort meant to operate at the clinical stages of drug development, even though the translation problem there is, theoretically, ‘easy’.
Other than us. Noetik is building virtual cells with the explicit goal of assisting with clinical-stage problems: identifying responders to drugs and refining patient inclusion criteria for trials. At the same time, we believe that the tools we create in this process will also have powerful applications in pivotal, high-risk areas of preclinical research, such as target selection, while remaining grounded in human-level data. All three will be discussed in this essay series.
How do we do this? Our view is simple: the shortest path to usefulness is not maximal simulation on unrealistic biology, but grounded observations into realistic biology. We built that foundation first. Every datapoint that trains our virtual cell models comes from human tumor resections: 77M cells across ~2,500 patients across a dozen+ cancers, with paired spatial transcriptomics, spatial proteomics, exomes, and H&E’s from each one collected in our lab. In total, this is easily one of the largest datasets of its kind. And not a single cell line. We strongly believe that this means the path from in-silico workflows to something clearly translatable is far more direct: human to human, rather than detouring through unrealistic animal or cell models.
That difference matters! In cancer, translation is the bottleneck. Drugs fail, not because they don’t work in preclinical settings, but because they don’t work in real human patients.
Using this human-derived tumor data, one of the virtual cell models we’ve created is ‘OCTO-VC’. This model is entirely trained on 1000-plex spatial transcriptomes, and its core task is deliberately prosaic: given the transcriptome of a few neighboring cells, reconstruct the “center cell” transcriptome—over every cell, in every tumor, for every patient. We released a (very long) post late last year discussing it in depth for those who are curious about the machine-learning details, alongside an online demo.
But what wasn’t discussed in that earlier post is how one can use models like this for clinically meaningful, non-trivial problems.
In this essay series, we hope to do exactly that, by showing three case studies of times where OCTO-VC was directly useful for our therapeutics team.
This is part 3, which will discuss how we would use the perturbation capabilities of the model to identify novel targets for T cell effector state modulation.
Virtual perturbations that shift T cell effector state in humans
Therapeutic Context:
Two particularly common lung cancer mutations you’ll often see people discussing are KRAS and STK11. KRAS is one of the most frequent oncogenic drivers (i.e. causes the cancer in the first place), whereas STK11 is a tumor suppressor gene whose inactivation disrupts cellular metabolism and immune signaling. And, while KRAS-mutant tumors are quite common, STK11 shows up alone less frequently, more-so appearing alongside KRAS.
Tumors with this genetic combination are often referred to, unsurprisingly, as ‘KRAS STK11’. And, when the two mutations do appear together, the combination produces a particularly aggressive biology: tumors that are metabolically rewired, immunologically “cold,” and broadly resistant to both standard chemotherapies and immune checkpoint blockade. As expected, the clinical data consistently show the impact of this on patients: significantly shorter lifespans.
As of today, there are no approved therapies that directly address the KRAS STK11 genotype. Patients are typically treated with the same immunotherapy regimens offered to the broader non-small cell lung cancer population: immune checkpoint blockades. While this often works fine in KRAS patients, the efficacy of this class of drugs is far worse for the KRAS STK11 patients. And, given that the latter group isn’t particularly rare, millions of patients are likely underserved.
Question:
Which therapeutic targets, if targeted, would help cancer patients with KRAS STK11 mutations?
What we found:
Well, perhaps we should first ask a simpler question: what exactly is the fundamental difference between KRAS and KRAS STK11 patients in cell-types most relevant to immunotherapy? KRAS patients, after all, respond well to immunotherapy, so they could be considered a model population for understanding what “good” looks like in terms of immune biology. Afterwards, we can move onto assessing what targets are most relevant to shifting KRAS STK11 tumors to have that particular phenotype.
For both of these, we leaned heavily on OCTO-VC’s ability to simulate cellular states.
First, to assess differences between the two population genotypes, we set up a ‘virtual CD8⁺ T cell simulation’. Here, we asked OCTO-VC to predict the “expected”, or virtual, CD8⁺ T cell in the genetic and microenvironmental context of each patient's tumor. And what we found is that one of the strongest differences in gene expression between KRAS and KRAS STK11 patients were a class of genes called granzymes, specifically GZMA and GZMK, which are known to be a practical readout of ‘CD8⁺ T-cell effector function’, the capacity for a T cell to kill cancer via cytotoxic mechanisms.
In the below plot, Gene A is GZMA and Gene B is GZMK. We’ll discuss in the next section why we believe these virtual cell predictions are a much better way to assess patient-level differences compared to the raw transcript values, but for now, we’ll move on.
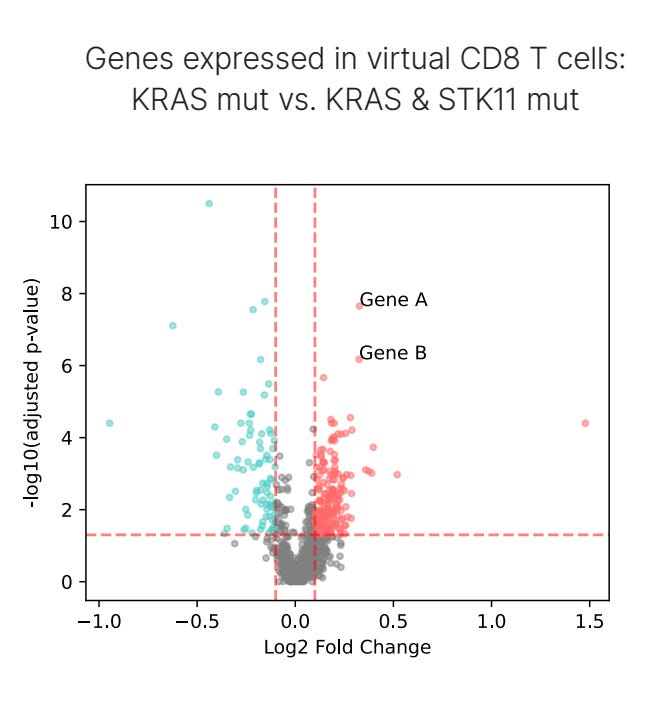
Step one completed, we’ve identified a therapeutically relevant difference between the two genotypes. Importantly, the marker does meet some sanity checks too. Granzyme expression has shown strong associations with response to PD-1/PD-L1 therapy in human tissue, clearly indicating that it is clinically meaningful for immunotherapy. So, one particular axis of improving the prospects of KRAS STK11 patients could be to simply find some way to increase granzyme levels.
But understanding the best ways to do this has been far from straightforward. Cytokine stimulation or blocking checkpoint molecules like TIGIT have all been shown in preclinical animal models to boost granzyme expression. Yet the current translational record is mixed: interventions that should theoretically raise granzyme levels often fail to yield durable tumor clearance in human clinical trials.
What’s going on here? Are granzymes the wrong lever to pull?
Perhaps, but there’s some reason to believe that some of the previous attempts to increase granzymes (in humans) did not, in fact, actually increase granzymes. After all, the molecular impact of at least one of those attempts seems to rely on entirely different mechanisms of action, ones that, empirically, ended up having no real patient benefit. The fundamental problem here may not be that granzymes aren’t worth modulating in humans, but rather, the targets that modulate them depend on the species. In other words, if you study mice only, you’re going to arrive at the wrong target.
After all, the structures of granzymes substantially differ between humans and mice. Broader than this is that the fact that immunity is a very, very species-specific topic. Consider inflammation, a close relative to our subject, and what a 2013 PNAS paper has to say about the role of mouse studies here (bolding added by me):
Murine models have been extensively used in recent decades to identify and test drug candidates for subsequent human trials. However, few of these human trials have shown success. The success rate is even worse for those trials in the field of inflammation, a condition present in many human diseases. To date, there have been nearly 150 clinical trials testing candidate agents intended to block the inflammatory response in critically ill patients, and every one of these trials failed.
All this to say: if we want to modulate granzymes, and come to useful conclusions about how to do so, we should work directly with human data. One way to do this (perhaps the only way to do it!) is to rely on OCTO-VC’s ability to perform virtual perturbations in real human data.
With the same computational framework as before—asking the model to predict virtual CD8⁺ T cells in a specific tumor microenvironment—we added one more step: knocking out a single gene across the tumor. From there, we ask how the virtual CD8⁺ T cell’s transcriptome would shift in response to that, comparing it to the baseline expected transcriptome of that cell type. We can do this systematically across thousands of genes to run a virtual screen. The knockout serves as a proxy for a drug, and the predicted impact on the virtual CD8⁺ T cell serves as a proxy for patient response. Of course, this impact is not at all guaranteed to be causal, merely strongly correlated and conditional on the spatial environment, but it can lead to useful hints.
We did exactly this perturbation across our KRAS STK11 patient cohort, searching for targets that consistently increased one of the granzymes, GZMA, expression in CD8⁺ T cells in real tumors. The virtual screen produced a clear signal: the top-scoring hit (Gene 20) was an adhesion protein, which we’ll call Target A.
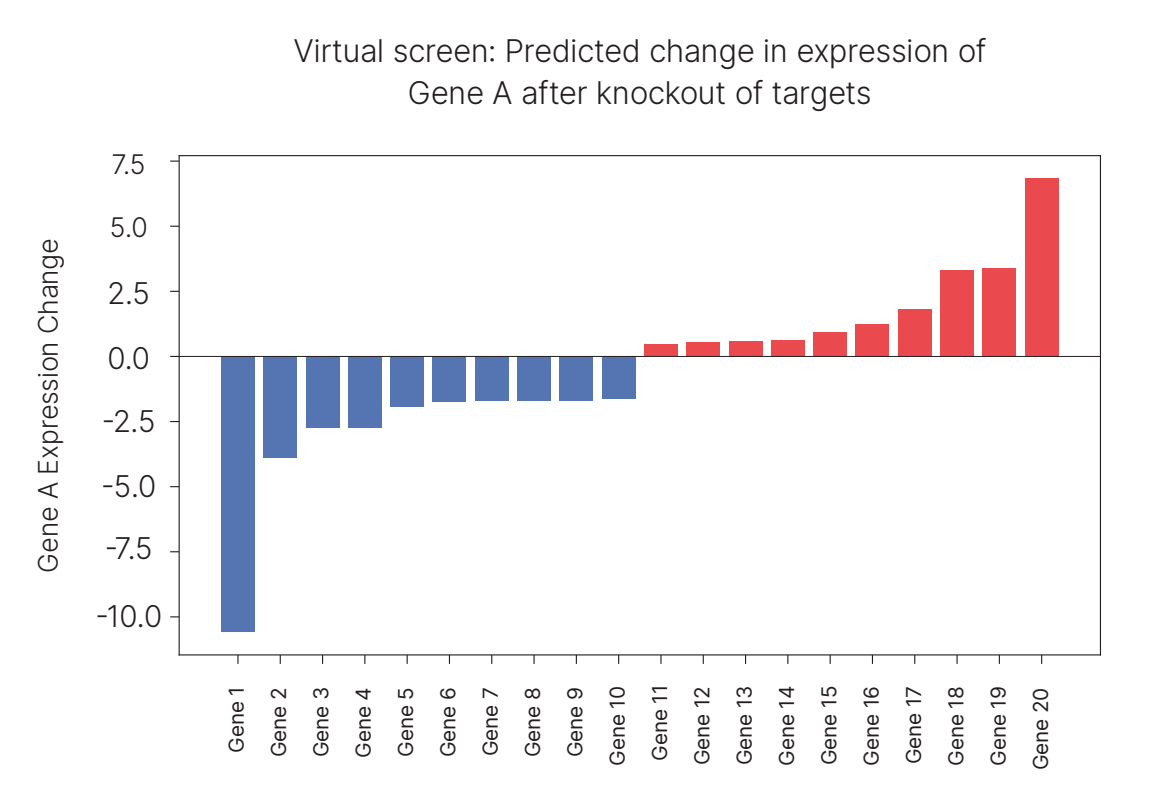
Target A is particularly intriguing because a study published only a few years ago showed that inhibiting this target (in co-cultured human tumors with T cells) leads to increased T cell expression of a granzyme. One nuance is that that papers granzyme studied GZMB, not GZMA, but the two can be quite correlated. But most compelling of all, beyond in-vitro results, is that there are two human cancer trials that have tested drugs meant to inhibit Target A!
How have these trials gone? It’s a mixed bag: patients responded decently in one trial, but not in the other. But both of them are using the exact same inclusion criteria: elevated levels of Target A. We strongly believe that this may have hurt both of the trial readouts.
Remember, inhibiting Target A in KRAS tumors is unlikely to yield immense benefits, since we suspect the primary mechanism-of-action of Target A is in in increasing granzyme activity, and those tumors already harbor abundant granzyme activity. In contrast, KRAS STK11 tumors, which have depressed granzyme levels, stand to gain the most from Target A inhibition. So, by enrolling patients purely on the basis of ‘high Target A expression’, the trials were almost certainly accidentally enriched for KRAS patients—by virtue of KRAS being found in 30% of all cancers, while KRAS STK11 are found in 10% of all cancers— inadvertently selecting a patient demographic least likely to respond to the drug.
Both of the trials, in other words, potentially stacked the deck against themselves. The correct strategy would have been to include KRAS STK11 status in the inclusion criteria, thereby focusing on patients with the greatest mechanistic rationale for benefit. But the trials did not do this and, as a result, the final efficacy readouts of the drug may be worse than it could’ve been.
Future Directions:
In one fell swoop, our virtual cell model uncovered not only a therapeutically relevant target, but also inclusion criteria on what patients it is most relevant for. Though there are already ongoing trials for this particular target, we strongly believe that the correct inclusion criteria for it are not being used.
Is there a principled way this could’ve been done without OCTO-VC?
For finding the granzyme difference between the two genotypes, theoretically yes, but practically no.
For finding target A, neither practically nor theoretically.
One, on the granzyme difference: though granzymes are known to be markers of CD8⁺ T cell effector function, their modulation in genetic subcontexts like KRAS STK11 has not, as far as we can tell, been systematically mapped. But even if you had collected the same spatial transcriptomics dataset we had, discovering this relationship without OCTO-VC would’ve been challenging. Why? Because raw transcript values are, generally speaking, untrustworthy. To assess the transcriptional differences between two cohorts based on raw genes, you would need CD8⁺ T cells to actually be present in sufficient numbers within the tumor microenvironment and ensure that those cells were captured with sufficiently-high resolution.
This is rarely the case! In most of our samples, even correctly tagging a cell as being a CD8⁺ T cell is difficult, to say nothing of their transcripts, which are often sparse, heterogeneous, and noisy, making it difficult to detect consistent patterns. Virtual cells, produced by OCTO-VC, solve this bottleneck by being able to reconstruct what a CD8⁺ T cell state would look like in that genetic and microenvironmental context; conditioned on the spatial transcriptomic environments the model has observed across millions of cells.
And two, on finding target A: even if you could extract a clean signal from the raw data, discovering targets that modulate the granzyme phenotype further would be largely intractable. The typical way people would study this further is via animal studies, and, as we’ve mentioned, there is a massive gulf between what mice immune systems tell you and what human immune systems tell you. The only way to reliably explore the area is via screening targets in a human, in vivo context, which necessitates the usage of virtual cell models like OCTO-VC to do it at any reasonable scale.
And though Target A was discovered without OCTO-VC, its discovery relied on cell culture data. The results of this coincidentally translated to humans, but, given how often cancer drugs fail, it’s a very expensive coin flip to make and not something we consider particularly principled.
These results are, to put it lightly, exciting. The history of cancer drug development has shown us time and time again that translation is the bottleneck. The problem has always been that what works in a mouse, or in a dish, rarely works in a patient. That’s it. Fixing this is how we make a dent in stopping the millions of lives that are lost to cancer every year. And we fix it by not being able to predict the results of a functional assay, or cell-line experiment, or mouse experiment. We fix it by trying to predict what happens when a human being with a real tumor gets treated. That’s the only question that matters. Everything else is a proxy, a bad proxy, one that has led to 90%+ of all cancer drugs failing during clinical trials.
We are not the first ones to claim that predictions like that are possible, but we believe that we are one of the first to show concrete evidence of it actually being done. And remember, the results we have today are the worst ones we’ll ever have. Each day, the practical utility of the model that fueled these results gets better and better, both as its underlying dataset grows richer and our understanding of how to best deploy it is refined.
The trajectory to us feels obvious; in time, models like OCTO-VC will become routine parts of how oncology as a field functions. In such a world, patients don’t waste precious time on ineffective treatments, entirely new targets that once seemed unworkable become viable options, and trial populations are enriched for the responders who stand to benefit the most. We have strong conviction that not only is this world possible, but that it is already beginning to emerge.
If any of this seems interesting, please reach out to info@noetik.ai to chat further.
If you’d like to read our prior case studies, here is Part 1 and Part 2.
Finally, if you’re curious to understand more ML-specific technical details about how the virtual CD8⁺ T cell’s actually work, we have an older post that discusses exactly that.